Download de presentatie
De presentatie wordt gedownload. Even geduld aub

1
Gezamenlijke studies Inez Bronsveld, longarts, UMC Utrecht
Els van der Wiel, research coordinator ErasmusMC-Sophia

2
Inhoud Kennismakingsrondje/vragen/verwachtingen
Wet en regelgeving bij wetenschappelijk onderzoek WMO-plichtig? GCP Infrastructuur per CF centrum nodig voor het doen van onderzoek Gezamenlijke studies

3
Wet- en regelgeving medisch wetenschappelijk onderzoek
Nationaal: Burgerlijk recht WOG WMO WBIG KWZ WBP WGBO Internationaal: Directives EMEA Clinical trial directive ( WMO) Data protection (WBP) Medicine (WOG) ICH GCP guidelines (Good Clinical Practice)
 Data protection (WBP) Medicine (WOG) ICH GCP guidelines (Good Clinical Practice)")
4
Regelgeving relaties

5
Welk onderzoek is WMO plichtig?
Onderzoek met mensen moet een medisch-ethische toets ondergaan als het valt onder de Wet medisch-wetenschappelijk onderzoek met mensen (WMO). Onderzoek valt onder de WMO als aan de volgende twee voorwaarden is voldaan: er is sprake van medisch-wetenschappelijk onderzoek personen worden aan handelingen onderworpen of hen worden gedragsregels opgelegd
. Onderzoek valt onder de WMO als aan de volgende twee voorwaarden is voldaan: er is sprake van medisch-wetenschappelijk onderzoek. personen worden aan handelingen onderworpen of hen worden gedragsregels opgelegd.")
6
In te dienen documenten
Te vinden op website CCMO of intranet van “eigen” METC A1: Aanbiedingsbrief toetsingscommissie (versie maart 2009) A1: Aanbiedingsbrief bevoegde instantie (versie maart 2009, alleen bij geneesmiddelenonderzoek) A3: Aanvraag EudraCT-nummer (alleen bij geneesmiddelenonderzoek) B1: ABR-formulier (online versie via ToetsingOnline) Ter informatie: Tekstversie ABR-formulier (met toelichting bij de vragen) B3: EudraCT aanvraagformulier (Application Form) (alleen bij geneesmiddelenonderzoek) C1: Model onderzoeksprotocol (toelichting) D2: Model IMPD (toelichting) (alleen bij geneesmiddelenonderzoek) E1/E2:Model informatiebrief (inclusief toestemmingsverklaringen, versie 3 november 2008) G1: Model verzekeringstekst proefpersoneninformatie (versie 3 november 2008) I2: Model lokale uitvoerbaarheidsverklaring (versie juni 2009) Verplicht bij multicenteronderzoek. Zie het thema multicenteronderzoek K3: CCMO model onderzoekscontract (Engels) (versie november 2008). Zie het nieuwsbericht
 A1: Aanbiedingsbrief bevoegde instantie (versie maart 2009, alleen bij geneesmiddelenonderzoek) A3: Aanvraag EudraCT-nummer (alleen bij geneesmiddelenonderzoek) B1: ABR-formulier (online versie via ToetsingOnline) Ter informatie: Tekstversie ABR-formulier (met toelichting bij de vragen) B3: EudraCT aanvraagformulier (Application Form) (alleen bij geneesmiddelenonderzoek) C1: Model onderzoeksprotocol (toelichting) D2: Model IMPD (toelichting) (alleen bij geneesmiddelenonderzoek) E1/E2:Model informatiebrief (inclusief toestemmingsverklaringen, versie 3 november 2008) G1: Model verzekeringstekst proefpersoneninformatie (versie 3 november 2008) I2: Model lokale uitvoerbaarheidsverklaring (versie juni 2009) Verplicht bij multicenteronderzoek. Zie het thema multicenteronderzoek K3: CCMO model onderzoekscontract (Engels) (versie november 2008). Zie het nieuwsbericht.")
8
U (als verrichter/sponsor) bent verplicht de volgende zaken aan de METC te melden: - Start- en einddatum - Wijzigingen onderzoeksdossier - SAE/onverwachte bijwerkingen - Voortgang - Voortijdige beëindiging onderzoek - Eindresultaten
 bent verplicht de volgende zaken aan de METC te melden: - Start- en einddatum - Wijzigingen onderzoeksdossier - SAE/onverwachte bijwerkingen - Voortgang - Voortijdige beëindiging onderzoek - Eindresultaten")
9
De beginselen van ICH GCP
2.1 Klinisch onderzoek moet worden uitgevoerd in overeenstemming met de ethische beginselen die hun oorsprong vinden in de Verklaring van Helsinki en die overeenstemmen met GCP en de relevante wettelijke vereisten. 2.2 Vóór de aanvang van een klinisch onderzoek moeten de te verwachten risico’s en ongemakken worden afgewogen tegen het te verwachten voordeel voor de individuele proefpersoon en de samenleving. Een klinisch onderzoek mag alleen worden opgezet en voortgezet als de te verwachten voordelen de risico’s rechtvaardigen. 2.3 De rechten, de veiligheid en het welzijn van de proefpersonen vormen de belangrijkste overwegingen en moeten prevaleren boven de belangen van wetenschap en samenleving. 2.4 De beschikbare pre-klinische en klinische informatie betreffende een onderzoeksproduct moet toereikend zijn om het onderzoeksvoorstel te onderbouwen. 2.5 Klinisch onderzoek moet wetenschappelijk verantwoord zijn en moet worden beschreven in een duidelijk en gedetailleerd protocol. 2.6 Een klinisch onderzoek moet worden uitgevoerd overeenkomstig het protocol waarop vooraf een positief oordeel is verkregen van een Medisch-ethische toetsingscommissie (METC). 2.7 De medische zorg voor en de medische beslissingen ten behoeve van proefpersonen moeten altijd vallen onder de verantwoordelijkheid van een bevoegd arts of, indien van toepassing, een bevoegd tandarts. 2.8 Elke persoon die betrokken is bij de uitvoering van een klinisch onderzoek moet door opleiding, training en ervaring gekwalificeerd zijn om zijn of haar respectievelijke taak/taken uit te voeren. 2.9 Voorafgaand aan deelname aan een klinisch onderzoek moet van elke proefpersoon een informed consent worden verkregen, die geheel vrijwillig is gegeven. 2.10 Alle informatie betreffende een klinisch onderzoek moet op zodanige wijze worden vastgelegd, behandeld en opgeborgen dat deze beschikbaar is voor nauwgezette rapportage, interpretatie en verificatie. 2.11 De vertrouwelijkheid van de documenten waarmee proefpersonen kunnen worden geïdentificeerd moet worden beschermd, waarbij de regels voor privacy en vertrouwelijke behandeling moeten worden nageleefd in overeenstemming met de relevante wettelijke vereisten. 2.12 Onderzoeksproducten moeten worden gefabriceerd, gehanteerd en opgeslagen in overeenstemming met de relevante Good Manufacturing Practice (GMP).Zij moeten worden gebruikt in overeenstemming met het goedgekeurde protocol. 2.13 Er moeten systemen worden ingevoerd met procedures die de kwaliteit van elk aspect van het onderzoek waarborgen.
. 2.7 De medische zorg voor en de medische beslissingen ten behoeve van proefpersonen moeten altijd vallen onder de verantwoordelijkheid van een bevoegd arts of, indien van toepassing, een bevoegd tandarts. 2.8 Elke persoon die betrokken is bij de uitvoering van een klinisch onderzoek moet door opleiding, training en ervaring gekwalificeerd zijn om zijn of haar respectievelijke taak/taken uit te voeren. 2.9 Voorafgaand aan deelname aan een klinisch onderzoek moet van elke proefpersoon een informed consent worden verkregen, die geheel vrijwillig is gegeven Alle informatie betreffende een klinisch onderzoek moet op zodanige wijze worden vastgelegd, behandeld en opgeborgen dat deze beschikbaar is voor nauwgezette rapportage, interpretatie en verificatie De vertrouwelijkheid van de documenten waarmee proefpersonen kunnen worden geïdentificeerd moet worden beschermd, waarbij de regels voor privacy en vertrouwelijke behandeling moeten worden nageleefd in overeenstemming met de relevante wettelijke vereisten Onderzoeksproducten moeten worden gefabriceerd, gehanteerd en opgeslagen in overeenstemming met de relevante Good Manufacturing Practice (GMP).Zij moeten worden gebruikt in overeenstemming met het goedgekeurde protocol Er moeten systemen worden ingevoerd met procedures die de kwaliteit van elk aspect van het onderzoek waarborgen.")
10
Infrastructuur per CF centrum
METC Juridische dienst Financiële dienst Voldoende patiënten Voldoende staf Klinische zorg volgens CBO richtlijnen Committment to research SOP’s aanwezig: algemene research en instelling specifiek Research personeel PI en RV/RC

11
Gezamenlijke studies Multi center nationaal
Multi center internationaal Nationaal, 1 onderzoekscentrum, meerdere patiënten vanuit andere centra Een naam voor de laatste!!! Intractief, geen ervaring mee graag meedenken

12
Multicenter nationaal
Ondersteuning vanuit de NCFS → Nederlands CF research netwerk Stuurgroep en werkgroep Doel: alle mensen met CF in NL kunnen deelnemen aan onderzoek hoogwaardige data verzamelen (kwaliteit ↑) waar mogelijk financiële ondersteuning bieden Stuurgroep ned cf research NETWERK en werkgroep researchcoordinatie verpleegkundigen
 waar mogelijk financiële ondersteuning bieden. Stuurgroep ned cf research NETWERK en werkgroep researchcoordinatie verpleegkundigen.")
13
Nederlands CF research netwerk
Stuurgroep ( 2 vertegenwoordigers per centrum, 2x per jaar vergaderen) Bevorderen van samenwerking Bepalen van beleid Prioriteiten van studies Waarborgen van kwaliteit van de uitvoering van de studies in alle centra Vanuit ieder CF centrum 2 vertegenwoordigers
 Bevorderen van samenwerking. Bepalen van beleid. Prioriteiten van studies. Waarborgen van kwaliteit van de uitvoering van de studies in alle centra. Vanuit ieder CF centrum 2 vertegenwoordigers.")
14
Nederlands CF research netwerk
Werkgroep research coordinatie (2 vertegenwoordigers per centrum, 2x per jaar vergaderen) Afstemmen voorbereiding, uitvoering en afsluiten studies Monitoren / verbeteren kwaliteit van de uitvoering Monitoren / verbeteren kwaliteit data verzamelen voor registratie
 Afstemmen voorbereiding, uitvoering en afsluiten studies. Monitoren / verbeteren kwaliteit van de uitvoering. Monitoren / verbeteren kwaliteit data verzamelen voor registratie.")
15
Projecten Nationale projecten Pseudomonasstammen Schimmelbank
Insuline project PREVEC studie Groeistudie CF registratie Internationale projecten Vertex809 PTC124 Tides studie Vanuit geld van de jubileum campagne van de NCFS zijn een aantal nationale projecten opgestart pseudomonas stammen In dit onderzoek zal worden onderzocht wat het effect is van infectie met clonale (gemakkelijk overdraagbaar) Pseudomonas aeruginosa stammen op de ziekte-ernst van patiënten met Cystic Fibrosis in Nederland. Schimmelbank Het doel van dit project is om te inventariseren welke schimmels bij de Nederlandse CF- patiënten voorkomen; of er verschillen bestaan in de frequentie, soorten en erfelijke eigenschappen (genotypes) van de schimmels per regio of per seizoen; of stammen binnen personen veranderen (muteren) of verschillende ongevoeligheid voor medicijnen (resistentie) ontwikkelen. Insuline project Het doel van dit onderzoeksproject is om aan te tonen dat het geven van lage hoev insuline de voedingstoestand van CF patienten verbetert. PREVEC studie Studie naar inflammatoire markers in condensaat, voor vroege herkenning van exacerbaties Groeistudie Verbeteren van de BMI bij CF patienten PTC 124 en Tides door inez besproken
 Pseudomonas aeruginosa stammen op de ziekte-ernst van patiënten met Cystic Fibrosis in Nederland. Schimmelbank. Het doel van dit project is om te inventariseren welke schimmels bij de Nederlandse CF- patiënten voorkomen; of er verschillen bestaan in de frequentie, soorten en erfelijke eigenschappen (genotypes) van de schimmels per regio of per seizoen; of stammen binnen personen veranderen (muteren) of verschillende ongevoeligheid voor medicijnen (resistentie) ontwikkelen. Insuline project. Het doel van dit onderzoeksproject is om aan te tonen dat het geven van lage hoev insuline de voedingstoestand van CF patienten verbetert. PREVEC studie. Studie naar inflammatoire markers in condensaat, voor vroege herkenning van exacerbaties. Groeistudie. Verbeteren van de BMI bij CF patienten. PTC 124 en Tides door inez besproken.")
16
Multicenter (inter)nationaal
Voordelen Grotere studie populatie Verbeteren expertise (NCFS/ECFS research netwerk ) Meer personeel Nadelen Verschil in wetgeving Verschil in werkwijze / SOP’s Meerdere indieningen bij EC’s Meer communicatie nodig Met name bij internat leidt dit tot langere aanloopfase ( vertealingen, tijdsverschillen)
 Meer personeel. Nadelen. Verschil in wetgeving. Verschil in werkwijze / SOP’s. Meerdere indieningen bij EC’s. Meer communicatie nodig. Met name bij internat leidt dit tot langere aanloopfase ( vertealingen, tijdsverschillen)")
17
ECFS-CTN Netwerk van expert CF centra
Samenwerking in klinische research Voor interant studies: ECFS

18
European CF Society Clinical Trial Network
Moeten aan bepaalde voorwaarden voldoen Begonnen met een klein aantal centra Opzet: Werken met elkaar SOPs ontwikkelen Interactie met sponsoren Doel om mee te doen in oa fase III studies waarvoor grote aantallen patienten nodig zijn netwerk wordt nu uitgebreid

19
Criteria voor deelnemende centra
Meer dan 75 pten in full care Bij voorkeur gedeelde kinder-/volwassenen centra Eerdere deelname aan fase II / III studies Geografie: iig uit 5 Europese landen en max 2 per land Clinical care volgens ECFS standaard Time commitment: protocol review, meetings, telefoon conferenties, educatie andere centra

20
Studies ECFS-CTN Competitive trials → snel handelen van belang, maar:
veel langere aanloopfase vertalen van alle documenten vragen metc, antwoorden metc, geadviseerde veranderingen van metc, patiënteninformatie alles loopt via de sponser elk centrum heeft eigen regeltjes tijdsverschillen tussen de deelnemende landen

21
Doel CTN meer uniformiteit in protocollen / SOP’s
Protocol ontwikkeling meer uniformiteit in protocollen / SOP’s meer gespecialiseerd personeel minder variabiliteit in data Recruitement van patienten door incidentie en complexheid van ziekte vaak moeilijk om genoeg inclusies te krijgen Nieuwe therapieën kunnen sneller geïmplementeerd worden Ontwikkelen van gezamenlijke database Samenwerking met farmacie Samenwerking met CFF-TDN Samen publiceren

22
Competenties Research verpleegkundige
Klinisch wetenschappelijk onderzoek beoordelen Klinisch wetenschappelijk onderzoek voorbereiden Toetsingsprocedures coördineren Onderzoek implementeren Onderzoekspersoon coördineren Onderzoek coördineren Data managen Onderzoek afsluiten en evalueren

23
VERTEX-809 Phase 2a trial evaluated the potential effect of an oral compound to improve trafficking of the defective CFTR protein (corrector) To evaluate the effect of VX-809 on biomarkers of CFTR activity homozygotes for the F508del: Spirometry (forced expiratory volume in 1 second [FEV1]) Sweat chloride concentration Nasal potential difference (NPD) Cystic Fibrosis Questionnaire-Revised (CFQ-R)
 Sweat chloride concentration. Nasal potential difference (NPD) Cystic Fibrosis Questionnaire-Revised (CFQ-R)")
24
2 cohort, placebo-controlled dose-escalation study
Group A: 25 mg, 50 mg, or placebo once daily Group B: 100 mg, 200 mg, or placebo once daily Dosing period: 28 days Study eligibility Aged 18 years and older Confirmed diagnosis of CF with F508del-CFTR mutation on both alleles

25
CFTR Correctors CFTR correctors aim to increase the delivery and amount of functional CFTR protein to the cell surface, resulting in improved ion transport ER VX-809 resulted from a high-throughput screening and medicinal chemistry optimization program to generate F508del-CFTR corrector compounds 25 Courtesy of JP Clancy

26
Enrolled and randomized
Study Design Placebo qd (n = 17) VX mg qd (n = 18) Enrolled and randomized (n = 89) Follow-up VX mg qd (n = 18) VX mg qd (n = 17) VX mg qd (n = 19) Day 0 7 14 21 28 +7 Subjects recruited in 2 Groups: Group A (placebo, VX mg, VX mg) Group B (placebo, VX mg, VX mg) Placebo group represents subjects from Group A and Group B Safety evaluation was done after enrollment of Group A Courtesy of JP Clancy
 VX mg qd (n = 18) Enrolled and randomized. (n = 89) Follow-up. VX mg qd (n = 18) VX mg qd (n = 17) VX mg qd (n = 19) Day Subjects recruited in 2 Groups: Group A (placebo, VX mg, VX mg) Group B (placebo, VX mg, VX mg) Placebo group represents subjects from Group A and Group B. Safety evaluation was done after enrollment of Group A. Courtesy of JP Clancy.")
27
CFTR-mediated Cl– Transport: Sweat Cl– biomarker
Change from baseline at Day 28 – difference vs placeboa a treatment effect analysis based on an Analysis of Covariance (ANCOVA) model Sweat Cl– responders change from baseline to Day 28 VX-809 Placebo (n=17) 100 mg (n=15) 200 mg (n=16) Responder 20,* n (%) 1 (6%) Responder 10,† n (%) 6 (40%)‡ 6 (38%)‡ 27 *20 mmol/L reduction vs baseline; †10 mmol/L reduction vs baseline; ‡P=0.02 vs placebo Courtesy of JP Clancy 27
 model. Sweat Cl– responders change from baseline to Day 28. VX-809. Placebo. (n=17) 100 mg (n=15) 200 mg (n=16) Responder 20,* n (%) 1 (6%) Responder 10,† n (%) 6 (40%)‡ 6 (38%)‡ 27. *20 mmol/L reduction vs baseline; †10 mmol/L reduction vs baseline; ‡P=0.02 vs placebo. Courtesy of JP Clancy. 27.")
28
Additional CFTR Biomarkers
CFTR-mediated Cl– transport by Nasal Potential Difference Zero Cl– + isoproterenol response Change from baseline at Day 28 – difference vs placeboa a treatment effect analysis based on an Analysis of Covariance (ANCOVA) model VX-809 Placebo* (n=10) 25 mg (n=8) 50 mg (n=7)† 100 mg (n=7) 200 mg (n=1) Subjects with B-band to C-band conversion from baseline to Day 28 CFTR maturation in Western blot analysis of rectal biopsy tissue *Combined data from Parts A and B †One subject enrolled in error was heterozygous for F508del; had C-band present at baseline and Day 28 28 Courtesy of JP Clancy 28
 model. VX-809. Placebo* (n=10) 25 mg. (n=8) 50 mg. (n=7)† 100 mg. (n=7) 200 mg. (n=1) Subjects with B-band to C-band conversion from baseline to Day 28. CFTR maturation in Western blot analysis of rectal biopsy tissue. *Combined data from Parts A and B. †One subject enrolled in error was heterozygous for F508del; had C-band present at baseline and Day Courtesy of JP Clancy. 28.")
29
Clinical Outcomes: FEV1 % predicted
Percent change from baseline at Day 28 – difference vs placeboa a treatment effect analysis based on an Analysis of Covariance (ANCOVA) model Subject-reported HRQOL – CFQ-R mean change from baseline at Day 28 VX-809 Domain Placebo (n=17) 25 mg (n=17) 50 mg (n=17) 100 mg (n=16) 200 mg (n=18) Respiratorya +4.53 -5.22† -6.32*† -1.29 +2.22 aMCID in respiratory domain is improvement ≥4 (Quittner AL et al Chest. 2009;135: ). *P<0.05 within-subject; †P<0.05 versus placebo 29 Courtesy of JP Clancy 29
 model. Subject-reported HRQOL – CFQ-R mean change from baseline at Day 28. VX-809. Domain. Placebo. (n=17) 25 mg (n=17) 50 mg (n=17) 100 mg (n=16) 200 mg (n=18) Respiratorya † -6.32*† aMCID in respiratory domain is improvement ≥4 (Quittner AL et al Chest. 2009;135: ). *P<0.05 within-subject; †P<0.05 versus placebo. 29. Courtesy of JP Clancy. 29.")
30
Conclusions Most adverse events were respiratory in nature and occurred in both treated and placebo groups Safety and tolerability profile in this study warrants the further clinical evaluation of VX-809 in patients with CF A statistically significant dose response to VX-809 in sweat Cl– concentration was observed Suggests that small molecule correction of F508del-CFTR in the sweat gland is possible in patients with CF No significant changes in other CFTR biomarkers or clinical outcome measures Future studies will be needed to explore the optimal dose of VX-809 and possible combination with a CFTR potentiator 30 Courtesy of JP Clancy

31
VERTEX-809 Beloop Centrale indiening bij de METC in het Erasmus MC Goedkeuring nam veel tijd in beslag Protocol ook al ingediend bij de METC in UMCU Op de dag van goedkeuring in Erasmus: Gebeld met METC Erasmus dat er goedkeuring was + PI Erasmus MC Bevestigingsbrief laten faxen Deze persoonlijk afgeleverd bij de METC UMCU Zelfde dag nog mail van goedkeuring METC in UMCU

32
Mail rondgegaan vanuit Vertex dat inclusie nog een paar dagen zou duren
5 patienten direct ingepland: weinig bedenktijd (eigenlijk 1wk) Deze doorgebeld aan Vertex Zijn geincludeerd op de laatste dagen voor sluiten van de inclusies Erasmus MC: helaas te laat met patienten includeren Leermomenten: Tijdspad belangrijk Genoeg tijd voor METC uittrekken, maar ook prompt reageren op hun berichten, niet laten liggen. Ook sponsor activeren Zelf actief achter correspondentie aan Centrale indiening bij voorkeur in centrum met snel METC proces
 Deze doorgebeld aan Vertex. Zijn geincludeerd op de laatste dagen voor sluiten van de inclusies. Erasmus MC: helaas te laat met patienten includeren. Leermomenten: Tijdspad belangrijk. Genoeg tijd voor METC uittrekken, maar ook prompt reageren op hun berichten, niet laten liggen. Ook sponsor activeren. Zelf actief achter correspondentie aan. Centrale indiening bij voorkeur in centrum met snel METC proces.")
33
Vertex-809 CRA veranderde tijdens studie
CRA wist weinig details van uitvoering NPD, zweettest, DNA, centraal lab, kortom van praktische uitvoering

34
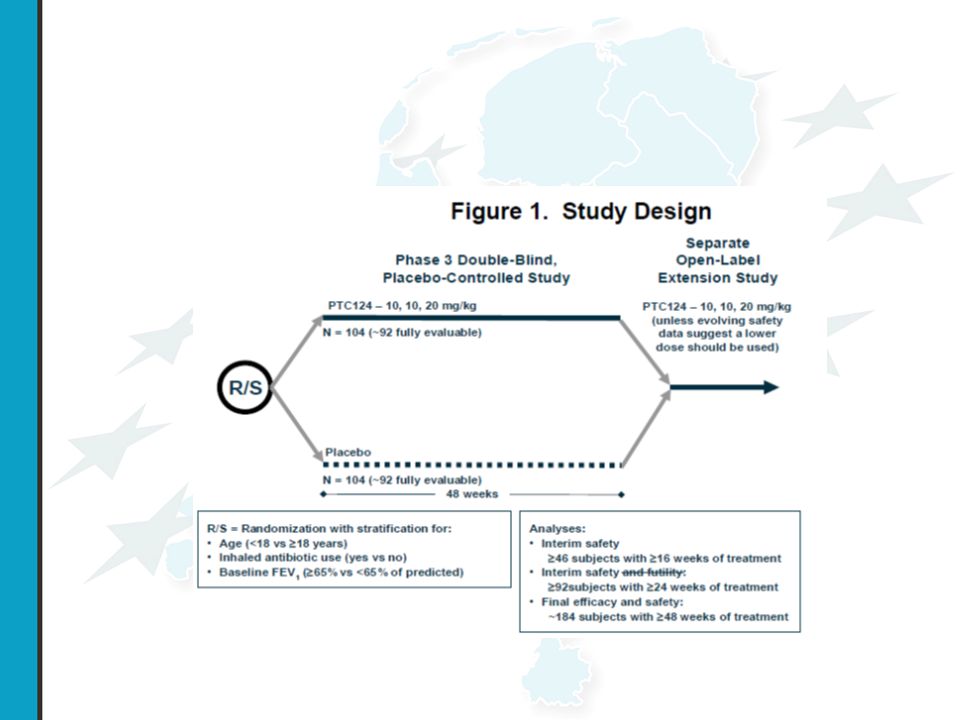
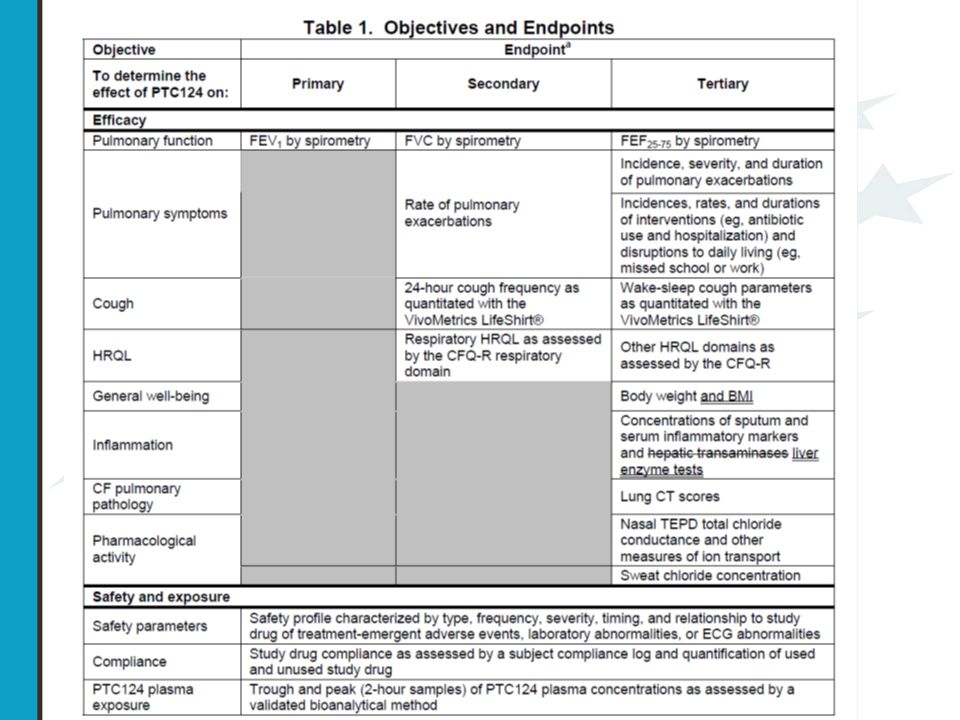
PTC-009 A Phase 3 Efficacy and Safety Study of PTC124 as an Oral Treatment for Nonsense-Mutation-Mediated Cystic Fibrosis CF patiënten met een stop codon mutatie

37
Beloop METC aanvraag Indiening Bureau Kwaliteitsborging Onderzoek
Goedkeuring? Dan METC indiening Afhankelijk van METC: Vergaderingen om de 2 weken Indiening 7-14 dagen van tevoren Contractjes met Radiologie, Laboratorium, Zweettestlab, Triallab, Apotheek Wachten Goedkeuring

39
PTC009 Bepalen juiste NPD protocol NPD instructie aan andere centra
Ondertussen heb je Investigator Meeting Bij deze studie: Bepalen juiste NPD protocol NPD instructie aan andere centra

40
Goedkeuring...en dan Dan pas mag je patienten gaan werven
Brief voor collega’s met in-/exclusie criteria Goedkeuring sponsor en METC Ondanks voorbereiding/vereenvoudiging vd documenten: dan nog bewerkelijk om te selecteren Drukke schema van artsen Research coordinatoren bieden uitkomst! Behandelaar zelf moet het aan de patient vragen

41
In-/exclusie criteria checken Alle gegevens uit andere ZH opvragen
Besluit tot deelname: In-/exclusie criteria checken Alle gegevens uit andere ZH opvragen Patient oproepen Screening

42
Deelname PTC009 Alle onderzoeken in UMCU Local Lab voor visites tussendoor met alleen bloedafname Groot voordeel voor de patient Mogelijke problemen: uitslagen van local lab worden niet doorgegeven/ moeilijk te achterhalen, bv nu urinesample Local Lab vraagt vergoeding: dit wordt geregeld door sponsor of tussenpersoon

43
Communicatie met Sponsor Tussenpersoon CRA Lead CRA Medical Monitor
Drug safety department Centraal lab voor analyses Centraal lab voor zweettest Centraal lab voor DNA analyse Insturen van NPD tracings Beoordeling van NPD tracing Centraal punt voor CT beoordeling Centraal punt voor ECG en longfunctie beoordeling resultaat als alles goed gaat: minstens 20 s per dag

44
AE/SAE in PTC-009 Exacerbatie Antibiotica kuren Abnormale bloedwaarden
Veel adverse events, bijvoorbeeld: Exacerbatie Antibiotica kuren Abnormale bloedwaarden Veranderde glucosewaarden Toename kortademigheid Buikpijn, droge mond, tintelingen, etc

45
Serious adverse events
Binnen 24 hrs melden Alle gegevens verzamelen tot in detail, zelfs als het uit Dax moet komen Labwaarden tijdens opname Start van event, start van serious criteria: dag, tijd Moeilijk te achterhalen Wanneer studie medicatie gestopt

46
Anurie Op vakantie....in Dax Controle de dag voor vakantie, maandag
Gordelroos...nog wat medicatie mee hiervoor, voorgeschreven door HA...is....? Flankpijn zaterdag Anurie woensdag, ging weer over Maar volgende dag wel naar SEH Dax NF: “goed”, echo goed. Blijkt later Kreat 123 Zondag terug: pijn nog steeds Direct naar SEH UMCU: NF gestoord, echo goed, geen bed Terug naar huis, opname maandag Urinesediment: eerst goed, dan veel leuco’s, dan geen leuco’s Nefroloog, Uroloog in consult: geen duidelijke oorzaak Blijkt pt ook nog 2 andere AB gehad te hebben, naast zithromax en acyclovir Tijdens opname: nogmaals kort moeite met plassen, ook verhoogd CRP Studiemedicatie / placebo? Oorzaak anurie ?

47
Patient motiveren 3x per dag medicatie innemen
Allemaal jonge mensen: school of baan Naast drukke dagindeling van alle reguliere medicatie Dagboekje bijhouden

48
Research coordinator Contact met METC over alle correspondentie
In UMCU: Contact met METC over alle correspondentie Contact met lab, radiologie, apotheek: binnen- en buitenland Na een studiedag: Faxen alle uitslagen Ontvangen alle uitslagen, bijhouden of alles is binnengekomen Investigator site file bijhouden (extra kast bestellen) Wekelijkse update van inclusie patienten, geplande screening, screenfailure Local lab inschakelen per patient Voorraden bijhouden en opschrijven Ruimte voor patient regelen Contact tussen WKZ en AZU Monitor visites begeleiden
 Wekelijkse update van inclusie patienten, geplande screening, screenfailure. Local lab inschakelen per patient. Voorraden bijhouden en opschrijven. Ruimte voor patient regelen. Contact tussen WKZ en AZU. Monitor visites begeleiden.")
49
PTC-009 voortgang Inclusie nu bijna voltooid
Patienten gaan door in de extension studie na 48 wk Open label, zelfde onderzoeken

50
TIDES studie Angst- en stemmingsklachten bij chronische aandoeningen
Hospital Anxiety and Depression Scale (HADS; Zigmond & Snaith, 1983) Center for Epidemiological Studies-Depression Scale (CES-D; Radloff, 1977) Cystic Fibrosis Questionnaire-Revised (CFQ-R; Quittner, Modi, Watrous, & Messer, 2000/2002) Ziekte Cognitie Lijst (ZCL; Evers & Kraaimaat, 1998) PROTOCOL: Voor polibezoek gesprek / uitleg / informed consent (PIF) Vragenlijsten invullen Na afloop bespreken uitslagen bespreken met patient Indien verhoogd: overleg over follow-up/ doorverwijzing Invullen in database best
 Center for Epidemiological Studies-Depression Scale (CES-D; Radloff, 1977) Cystic Fibrosis Questionnaire-Revised (CFQ-R; Quittner, Modi, Watrous, & Messer, 2000/2002) Ziekte Cognitie Lijst (ZCL; Evers & Kraaimaat, 1998) PROTOCOL: Voor polibezoek gesprek / uitleg / informed consent (PIF) Vragenlijsten invullen. Na afloop bespreken uitslagen bespreken met patient. Indien verhoogd: overleg over follow-up/ doorverwijzing. Invullen in database best.")
51
Eerste terugkoppeling TIDES
Lijkt eenvoudig Veel werk: Uitleg patient Vragenlijsten doornemen Resultaat met patient bespreken Invoeren in database

52
Gezamenlijke studies 1 studiecentrum, patienten vanuit meerdere CF centra
Doel Mogelijkheid tot deelname aan medisch wetenschappelijk onderzoek, nationaal of internationaal, voor elke CF patiënt binnen Nederland onafhankelijk van het centrum waar de patiënt behandeld wordt of waar het onderzoek uitgevoerd wordt. Is dus nationale studie, loopt via NCFS netwerk! Brengt andere logistiek met zich mee

53
Aandachtspunten mbt wet- en regelgeving
Toestemming sponsor. Alle studieprocedures in het CF onderzoekscentrum. Alleen het onderzoekcentrum heeft een contract ( en bijbehorende geheimhoudingsplicht) met de sponsor. Uitleg Patiënten Informatie Formulier. Patiënt/ouders moeten schriftelijk toestemming geven gegevens van hem/haar van het “eigen” CF centrum naar het onderzoekcentrum klinisch relevante data, verzameld tijdens deelname aan het onderzoek, naar eigen zorgcentrum Toestemming schriftelijk voor alle procedures! Evt geheimh verkl met andere centra, Personeel op trialstafflist PIF alleen door personen die op trial st list staan Schrift toest ouders / patient in status!
 met de sponsor. Uitleg Patiënten Informatie Formulier. Patiënt/ouders moeten schriftelijk toestemming geven. gegevens van hem/haar van het eigen CF centrum naar het onderzoekcentrum. klinisch relevante data, verzameld tijdens deelname aan het onderzoek, naar eigen zorgcentrum. Toestemming schriftelijk voor alle procedures! Evt geheimh verkl met andere centra, Personeel op trialstafflist. PIF alleen door personen die op trial st list staan. Schrift toest ouders / patient in status!")
54
Praktische uitvoering
Inlichten andere centra Patienten screenen en werven Gegevens overdragen SAE’s en AE’s Graag interactief, niemand heeft hier nog veel ervaring mee, graag meedenken

55
Inlichten andere centra
Algemene beschrijving van studie, patient direct doorsturen naar OC Bij toestemming sponsor Procedures schriftelijk vastleggen Geheimhoudingsverklaringverklaring, op trial staff list protocol en PIF naar andere centra Overleg studie in NCFS netwerk Algemene beschrijving van studie, patient direct doorsturen naar OC Bij toestemming sponsor Procedures schriftelijk vastleggen Geheimhoudingsverklaringverklaring, op trial staff list protocol en PIF naar andere centra Overleg studie in NCFS netwerk

56
Werving patienten Poster in wachtkamer/ longfunctie kamer
Bij toestemming sponsor Geheimhoudingsverklaringverklaring, op trial staff list protocol en PIF naar andere centra RV screent, arts licht patient in Overleg OC en BC Poster in wachtkamer/ long functie kamer Bij toestemming sponsor Geheimhoudingsverklaringverklaring, op trial staff list protocol en PIF naar andere centra RV screent, arts licht patient in Overleg OC en BC Werven:Werving van patiënten voor een onderzoek Het onderzoekcentrum zal alleen passief patiënten van andere centra kunnen werven. Bijvoorbeeld door middel van posters met informatie over het onderzoek in de wachtkamers, via de website van het centrum of via de NCFS. De patiënt/ouder kan dan zelf direct contact opnemen met het onderzoekcentrum. Een tweede optie is dat de sponsor het personeel van de centra waar het onderzoek niet uitgevoerd wordt, toestemming geeft tot inzage in het protocol zodat zij het onderzoek kunnen uitleggen aan de patiënten/ouders. Het personeel van deze centra zal dan een geheimhoudingsverklaring moeten tekenen met de sponsor. Diegenen die het onderzoek aan de patiënten/ouders uitleggen dienen op de trialstaf list van het studiecentrum te staan.

57
Gegevensoverdracht Pendelstatus ziektegeschiedenis huidige medicatie
longfunctiemetingen, sputumuitslagen enz. korte omschrijving van het onderzoek, het visiteschema, de toegestane co-medicatie, contactpersonen van het onderzoekcentrum contactpersonen van het zorgcentrum. Pendelstatus ziektegeschiedenis huidige medicatie longfunctiemetingen, sputumuitslagen enz. korte omschrijving van het onderzoek, het visiteschema, de toegestane co-medicatie, contactpersonen van het onderzoekcentrum contactpersonen van het zorgcentrum. Gegevensoverdracht De patiënt, die deelneemt aan een onderzoek, zal voor de studiebezoeken dus naar het onderzoekcentrum gaan. Voor de reguliere zorg blijft hij/zij onder controle in het eigen CF centrum. De patiënt krijgt, voor de duur van het onderzoek, een zogenaamde “pendelstatus”, hierin zal alle relevante informatie verzameld worden, de patiënt/ouders zijn zelf verantwoordelijk voor het meenemen van deze status bij bezoeken aan onderzoek – en zorgcentrum. In deze status zullen, afhankelijk van het protocol, meerdere kopieën aanwezig zijn o.a. van de ziektegeschiedenis en huidige medicatie van de patiënt, longfunctiemetingen en sputumuitslagen. De kopieën van alle brondocumenten (bijv. LF, genetische mutaties, microbiologie), dienen voorzien te zijn van handtekening (en datum) van de behandelend arts. Verder aanwezig in deze pendelstatus: korte omschrijving van het onderzoek, het visiteschema, de toegestane co-medicatie, de contactpersonen van het onderzoekcentrum en de contactpersonen van het zorgcentrum. De medewerkers van het zorgcentrum dienen te allen tijde op de hoogte te zijn van deelname anders kan dit problemen opleveren voor de behandeling van de patiënt zowel als voor de voortgang van de studie!! (onjuiste inschatting problemen, keuze medicatie enz).
, dienen voorzien te zijn van handtekening (en datum) van de behandelend arts. Verder aanwezig in deze pendelstatus: korte omschrijving van het onderzoek, het visiteschema, de toegestane co-medicatie, de contactpersonen van het onderzoekcentrum en de contactpersonen van het zorgcentrum. De medewerkers van het zorgcentrum dienen te allen tijde op de hoogte te zijn van deelname anders kan dit problemen opleveren voor de behandeling van de patiënt zowel als voor de voortgang van de studie!! (onjuiste inschatting problemen, keuze medicatie enz).")
58
SAE’s en AE’s Bij problemen gaat patient naar eigen centrum
Bij AE is patient “eigen bron” Bij SAE binnen 24u overleg BC en OC Cave co medicatie (pendelstatus) Bij problemen gaat patient naar eigen centrum Bij AE is patient “eigen bron” Bij SAE binnen 24u overleg BC en OC Cave co medicatie (pendelstatus) Vast leggen!!! Hoe te handelen Hoe te handelen bij bijv. exacerbaties of andere (Serious) Adverse Events? Bij een exacerbatie gaat de patiënt naar het eigen zorgcentrum en de gegevens worden gedocumenteerd in de pendelstatus. Er kan ook gekozen worden voor de optie dat de patiënt zelf vertelt wat er is gebeurd. Het (opgeschreven) verhaal van de patiënt functioneert dan als brondocument. Per onderzoek zal steeds overleg nodig zijn hoe een en ander gedocumenteerd en uitgevoerd moet worden
 Bij problemen gaat patient naar eigen centrum. Bij AE is patient eigen bron Bij SAE binnen 24u overleg BC en OC. Cave co medicatie (pendelstatus) Vast leggen!!! Hoe te handelen. Hoe te handelen bij bijv. exacerbaties of andere (Serious) Adverse Events Bij een exacerbatie gaat de patiënt naar het eigen zorgcentrum en de gegevens worden gedocumenteerd in de pendelstatus. Er kan ook gekozen worden voor de optie dat de patiënt zelf vertelt wat er is gebeurd. Het (opgeschreven) verhaal van de patiënt functioneert dan als brondocument. Per onderzoek zal steeds overleg nodig zijn hoe een en ander gedocumenteerd en uitgevoerd moet worden.")
Verwante presentaties