Download de presentatie
De presentatie wordt gedownload. Even geduld aub

1
Laboratoriumhematologie in de pediatrie
Jan Philippé UZ Gent - UGent

2
Referentiewaarden Hgb MCV Telling MCH hct Geboorte 14.5-22.0 95-125
31-37 46-60 1e week 90-120 42-64 2e week 86-120 28-40 39-63 1e maand 85-120 35-55 2e maand 80-115 26-34 30-42 3-6 maand 75-105 25-35 0.5-2 jaar 70-86 23-31 2-6 jaar 73-85 24-30 33-42 6-12 jaar 77-95 25-33 35-45

3
Referentiewaarden Hgb
Prematuur g g 2e week 1e maand 2e maand 3e maand 4e maand 5e maand 6e maand

4
Referentiewaarden Reticulocyten % X 109/L Erytroblasten Navelstreng
3-7 0-1.0 Dag 1 0-0.5 Dag 3 1-3 50-150 0-0.01 Dag 7 0.1-2 10-100 > 1 week

5
Referentiewaarden aantal immature RBC op de eerste geboortedag na verschillende zwangerschapsperioden Aantal weken zwschp Reticulocyten % Erytroblasten X 109/L 23-25 5-10 26-30 31-35 3-10 36-37 3-7 A term

6
Referentiewaarden Hgb F (%) Hgb A2 (%) 1-7 dagen 61-80 2 weken 66-81
1 maand 46-67 2 maand 29-61 3 maand 15-56 4 maand 9.4-29 5 maand 2.3-22 6 maand 2.7-13 8 maand 2.3-12 10 maand 13-20 maand 21-24 maand

7
Referentiewaarden IJzer µmol/L IBC Ferritine µg/L 2 weken 11-36 18-50
Transferrine sat % Ferritine µg/L 2 weken 11-36 18-50 30-39 25-200 1 maand 10-31 20-52 35-94 2 maand 3-29 24-64 21-63 50-200 4 maand 40-68 7-53 20-200 6 maand 5-24 40-76 10-43 7-142 0.5-4 jaar 5-25 48-79 10-40 5-10 jaar 5-30 43-91 10-45

8
Referentiewaarden Haptoglobine (mg/dL) Navelstreng 1-7 d 0-41 1-4 w
1-7 d 0-41 1-4 w 0-45 1-3 m 41-95 3-6 m 64-134 6-12 m 43-160 1-5 j 51-160 5-10 j 62-186 > 10j 41-165

9
Referentiewaarden Totaal aant. neutro’s 0-60 h 2.9-14.5 X 109/L
5-28 d 1-6 m 0.5-8 j 8-16 j Immature neutro’s 0-1.4 0-0.6 0-0.5 Imm/tot neutro’s < 0.16 < 0.13 < 0.12 Imm/mat ratio 0-1 m ≤ 0.3

10
Referentiewaarden Lymfocyten Geboorte 2.0-11.0 X 109/L 12 h 24 h
1-2 w 1 m 6 m 1 j 2 j 4 j 6 j 8 j 10 j 16 j

11
Referentiewaarden (BM)
% van ANC 0-1 maand 1m – 1 j 1j – 7j 7j -14 j Blasten 0-4 0-3 Promyelo’s 0-4.5 Myelo’s 8-25 Meta+staven 15-35 10-30 Segmenten 5-30 Eo’s 0-7 0-5 1-9 Baso’s 0-0.8 Erytroblasten 6-38 M/E ratio Lymfocyten 5-60 15-45 5-35 plasmacellen 0-0.05

12
Microcytaire anemie bij het kind
Fe deficiëntie Chronische aandoening Thalass. minor Hgb (g/dL) Tot 2 Meestal > 7 Meestal > 9 MCV (fL) Kan < 50 Meestal > 50 Retics ↑ (No voor Rcorr) N of ↑ RDW ↑ RBC morfologie + + (rel. voor de anemie) Dimorfe RBC Frequent Basof. stippeling Zelden Variabel Aantal RBC Normaal tot ↑ MCV / RBC Meestal > 13 Meestal < 13 Ferritine / Fe / No tot ↑ / No/No TIBC N Tf saturatie N tot Respons op Fe per os Goed Zwak geen BM ijzerreserve N tot ↑ Sideroblasten (beperkt bruikbaar)
 Tot 2. Meestal > 7. Meestal > 9. MCV (fL) Kan < 50. Meestal > 50. Retics. ↑ (No voor Rcorr) N of ↑ RDW. ↑ RBC morfologie. + + (rel. voor de anemie) Dimorfe RBC. Frequent. Basof. stippeling. Zelden. Variabel. Aantal RBC. Normaal tot ↑ MCV / RBC. Meestal > 13. Meestal < 13. Ferritine / Fe. / No tot ↑ / No/No. TIBC. N. Tf saturatie. N tot Respons op Fe per os. Goed. Zwak. geen. BM ijzerreserve. N tot ↑ Sideroblasten. (beperkt bruikbaar)")
13
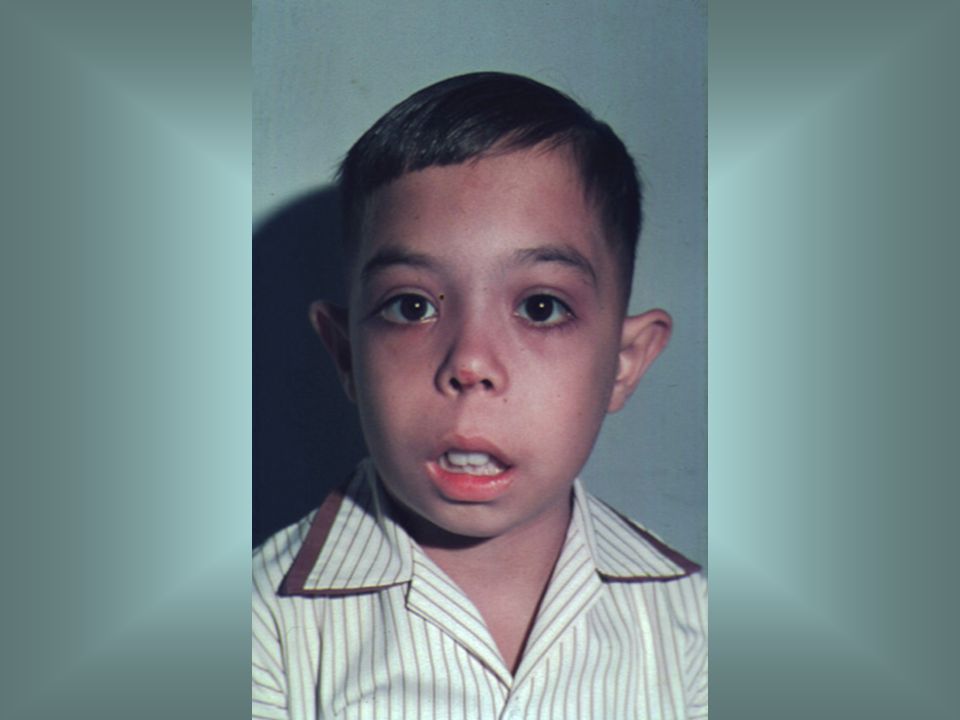
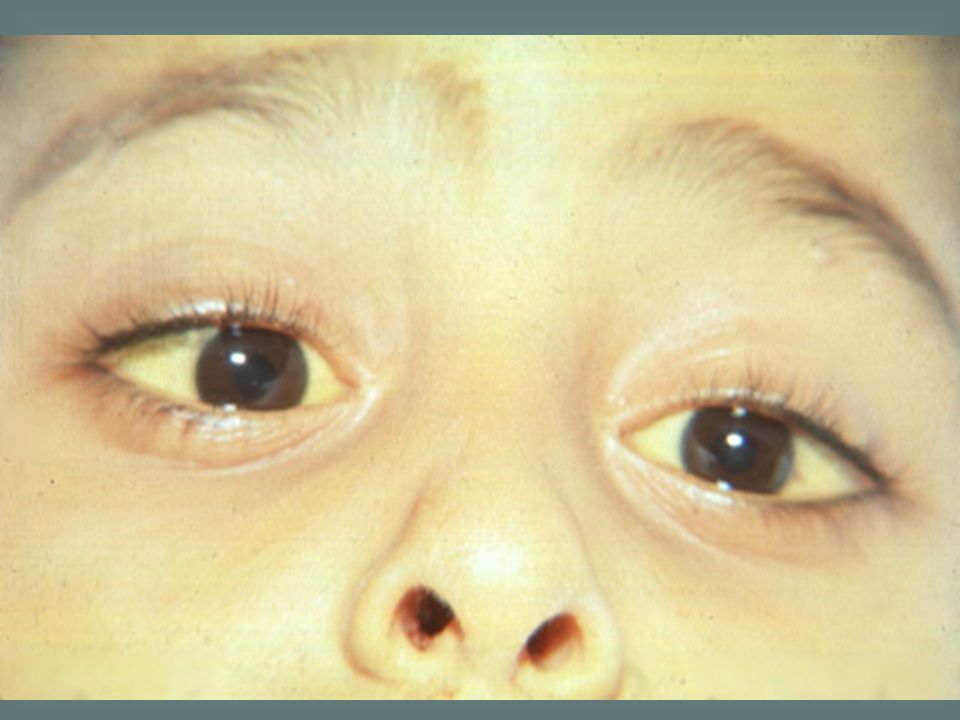
Congenital sideroblastic anaemia is a rare inherited abnormality in haem synthesis that is characterized by the presence of ring sideroblasts in the bone marrow. The blood film is characteristically dimorphic with a mixture of hypochromic microcytes and normocytic, normochromic cells. As in this case, there may also be poikilocytosis.

14
Most types of congenital sideroblastic anaemia are caused by a defect in haem synthesis, specifically a mutation in the ala synthase gene. Iron is taken up into the developing red cell but because of the lack of haem cannot be incorporated into haemoglobin. Iron is deposited in the mitochondria as haemosiderin and is apparent as a ring of iron-containing granules around the nucleus. Over 10% of NRBC are sideroblasts. DD with plumbism (acquired).
.")
15
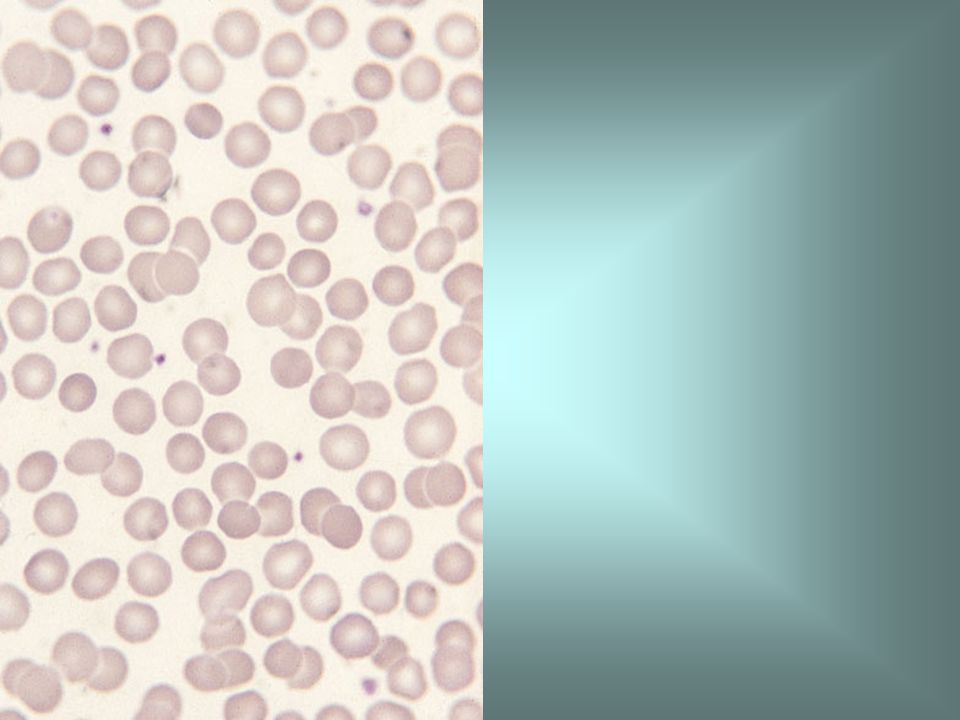
α thalassaemia trait means that there is loss of one or two of the normal complement of four α genes. This film, from an individual with α thalassaemia trait attributable to loss of two α genes, shows hypochromia and microcytosis. The diagnosis of α thalassaemia trait is difficult since it requires DNA analysis. Usually it is a presumptive diagnosis only, when no other explanation can be found for microcytic red cells.

16
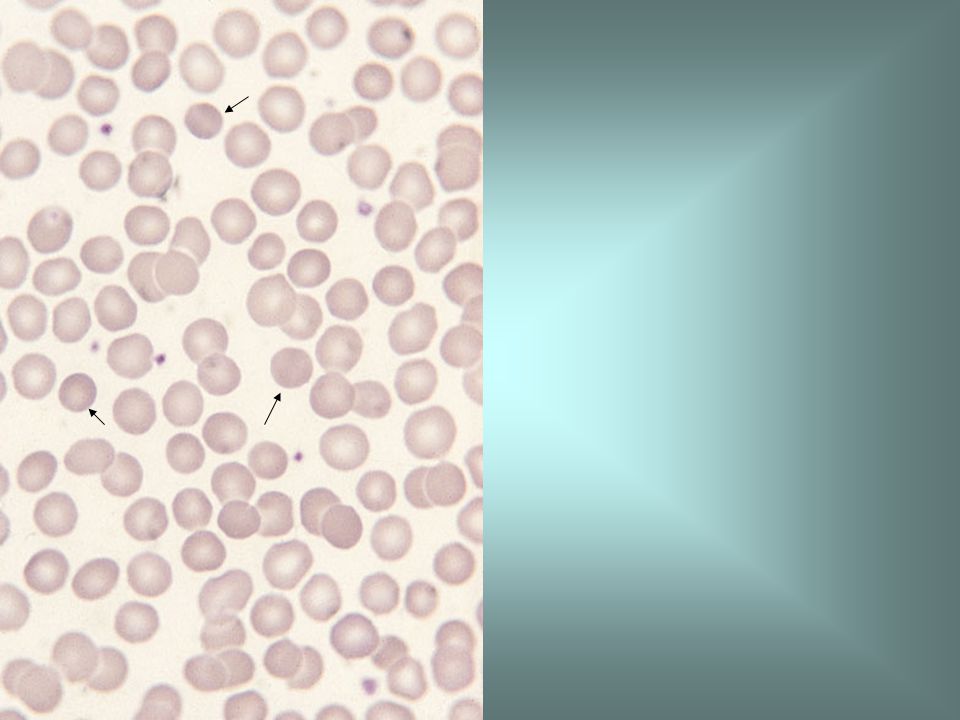
Haemoglobin H disease is a thalassaemic disorder with a marked reduction in a chain synthesis, usually caused by deletion of three of the four α genes. The blood film in haemoglobin H disease shows marked anisocytosis, poikilocytosis and hypochromia. The reticulocyte count is increased. The diagnosis of haemoglobin H disease is confirmed by demonstration of haemoglobin H on haemoglobin electrophoresis and on a haemoglobin H preparation.

17
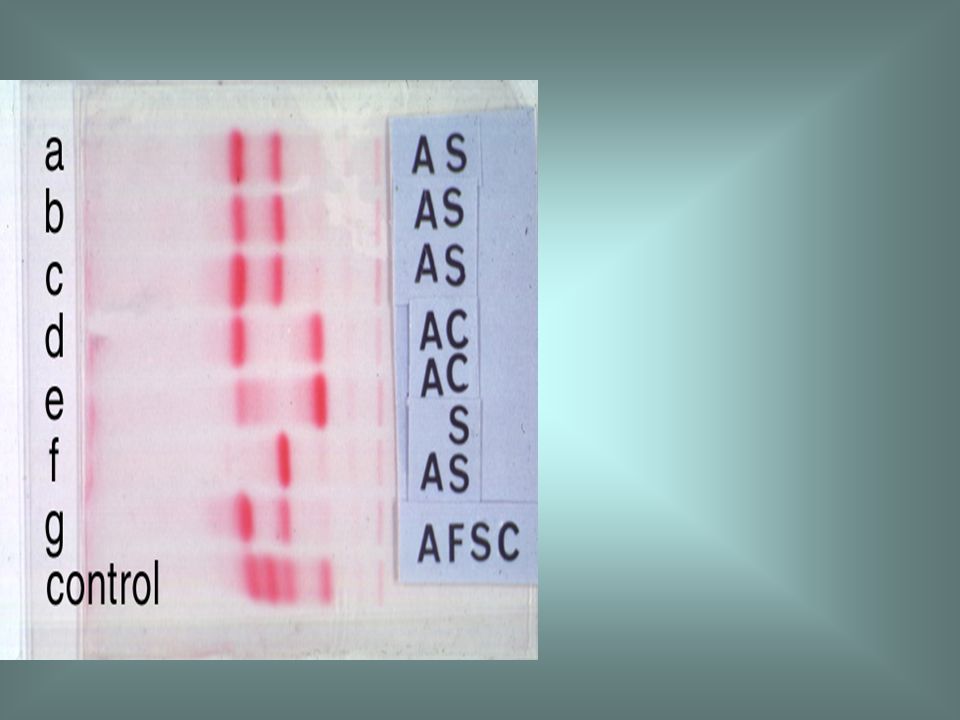
Haemoglobin electrophoresis in a patient with haemoglobin H disease (samples b and c) showing a minor fast band which is haemoglobin H. In haemoglobin H disease the percentage of the abnormal haemoglobin varies from 2 to 40%.

18
A haemoglobin H preparation shows that patients with haemoglobin H disease have a significant proportion of cells containing haemoglobin H inclusions [blue arrow]. These are small pale blue inclusions distributed evenly through a red cell, giving an appearance which has been compared to a golf ball. Increased numbers of reticulocytes [red arrows] are also apparent.
![A haemoglobin H preparation shows that patients with haemoglobin H disease have a significant proportion of cells containing haemoglobin H inclusions [blue arrow].](http://slideplayer.nl/slide/2028533/8/images/18/A+haemoglobin+H+preparation+shows+that+patients+with+haemoglobin+H+disease+have+a+significant+proportion+of+cells+containing+haemoglobin+H+inclusions+%5Bblue+arrow%5D..jpg "These are small pale blue inclusions distributed evenly through a red cell, giving an appearance which has been compared to a golf ball. Increased numbers of reticulocytes [red arrows] are also apparent..")
19
Β thalassaemia trait or β thalassaemia heterozygosity means that an individual has inherited a β thalassaemia gene from one parent and a normal β gene from the other parent. This film from a healthy person with β thalassaemia trait shows minimal morphological abnormality. There is microcytosis, slight hypochromia and slight poikilocytosis. Such cases are more readily suspected from the characteristic red cell indices than from the subtle abnormalities on the blood film.

20
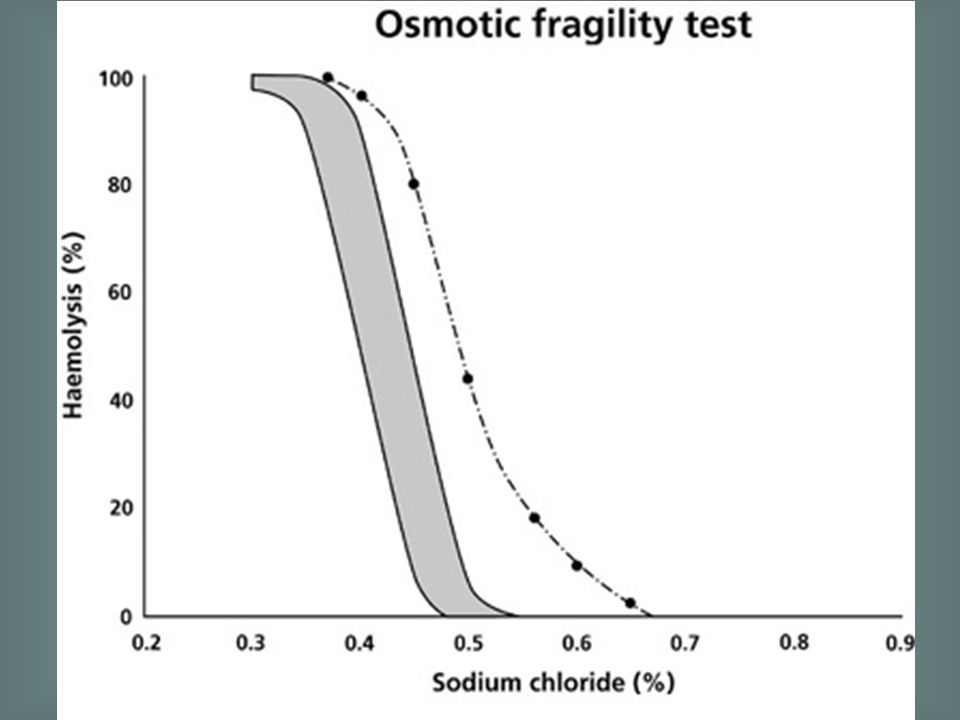
HbA2 > 3.5% in heterozygous β thalassaemia
Although the diagnosis of β thalassaemia trait can be suspected from the blood film, red cell indices and ethnic origin, definitive diagnosis requires measurement of the haemoglobin A2 percentage. This is elevated in β thalassaemia trait, as shown by this densitometric trace of an electrophoretic strip, whereas it is normal or reduced in α thalassaemia trait and iron deficiency anaemia. HbA2 > 3.5% in heterozygous β thalassaemia

22
β thalassaemia major is the name given to β thalassaemia of sufficient severity to require blood transfusion to maintain life. Anaemia becomes apparent at 3-6 months. This blood film is from a patient already receiving blood transfusions. It is therefore dimorphic with a mixture of the patient's thalassaemic red cells and normal donor cells. The patient's own red cells show hypochromia, target cell formation and Pappenheimer bodies [red arrow]. One red cell contains an inclusion that represents an α chain inclusion [blue arrow]. There are three NRBC. In β thalassaemia major Hb F > 80%

23
a siderotic granule (the equivalent of a Pappenheimer body) demonstrated with an iron stain
 demonstrated with an iron stain")
24
DD aangeboren hemolytische anemie
SCA/hemoglobine C Hemoglobine C Hemoglobine E hemoglobinopathieën Sikkelcelanemie Erfelijke sferocytose Erfelijke elliptocytose Erfelijke stomatocytose Pyruvaat kinase deficiëntie G-6-PDH deficiëntie (meest frequente oorzaak)
")
25
Haemoglobin electrophoresis on cellulose acetate at alkaline pH in haemoglobin C trait (strip d and e). At alkaline pH the findings in haemoglobin C trait and haemoglobin E trait are similar. If haemoglobin electrophoresis is also performed on agarose at acid pH the distinction is easy since haemoglobin E then has the same mobility as haemoglobin A whereas haemoglobin C does not.

26
Blood film in haemoglobin C disease showing target cells and irregularly contracted cells.
These two features are typical of haemoglobin C homozygosity. There may also be microcytosis. Rare haemoglobin C crystals may be seen.

29
The first haemoglobinopathy recognized was sickle cell anaemia, resulting from homozygosity for the HbS gene. -sickle cell (red arrow), -several boat-shaped cells (blue arrows) -and a Howell-Jolly body. -Hgb is decreased (7-10 g/dL) -RBC and Hct are similarly reduced -MCV and MCH are reduced in some patients. -RDW and reticulocyte count are increased. As hyposplenism develops there is a tendency for the neutrophil, lymphocyte and platelet counts to rise. The WBC and neutrophil count may rise further during sickle cell crisis.
, -several boat-shaped cells (blue arrows) -and a Howell-Jolly body. -Hgb is decreased (7-10 g/dL) -RBC and Hct are similarly reduced. -MCV and MCH are reduced in some patients. -RDW and reticulocyte count are increased. As hyposplenism develops there is a tendency for the neutrophil, lymphocyte and platelet counts to rise. The WBC and neutrophil count may rise further during sickle cell crisis.")
31
Blood film in sickle cell trait showing target cells and mild microcytosis. It should be noted that the blood film is often normal in sickle cell trait.

35
Hereditary elliptocytosis is a heterogeneous group of congenital haemolytic anaemias consequent on an inherited abnormality of the red cell membrane. Most cases are caused by a mutation in either the a or the b spectrin gene. Inheritance is usually autosomal dominant. Most patients have compensated haemolysis. Some have a mild, moderate or severe haemolytic anaemia.

36
Blood film in hereditary stomatocytosis showing basophilic stippling and numerous stomatocytes.
Hereditary stomatocytosis is a heterogeneous group of disorders resulting from an inherited abnormality of the red cell membrane. Inheritance is usually autosomal dominant. There may be compensated haemolysis or mild, moderate or severe haemolytic anaemia. Cation flux across the membrane may be abnormal. The film sometimes shows target cells, in addition to stomatocytes. Another inherited cause of stomatocytosis is Rh null disease so that Rhesus typing is indicated in congenital haemolytic anaemia associated with stomatocytosis.

37
The commonest cause of congenital haemolytic anaemia is glucose-6-phosphate dehydrogenase (G6PD) deficiency which affects millions of people world-wide. Most individuals with G6PD deficiency suffer only intermittent haemolysis. The blood film then shows irregularly contracted cells [deep red arrows] and sometimes hemighosts [deep blue arrow] in which all the haemoglobin appears to have retracted to one side of the erythrocyte. It has an X-linked recessive inheritance so occurs mainly in males. In high incidence areas female homozygotes occur and have the same features as male hemizygotes. Haemolysis is precipitated by infection and exposure to exogenous oxidants such as broad beans, naphthalene and certain drugs. Between acute haemolytic episodes the blood film is normal.
![The commonest cause of congenital haemolytic anaemia is glucose-6-phosphate dehydrogenase (G6PD) deficiency which affects millions of people world-wide. Most individuals with G6PD deficiency suffer only intermittent haemolysis. The blood film then shows irregularly contracted cells [deep red arrows] and sometimes hemighosts [deep blue arrow] in which all the haemoglobin appears to have retracted to one side of the erythrocyte.](http://slideplayer.nl/slide/2028533/8/images/37/The+commonest+cause+of+congenital+haemolytic+anaemia+is+glucose-6-phosphate+dehydrogenase+%28G6PD%29+deficiency+which+affects+millions+of+people+world-wide.+Most+individuals+with+G6PD+deficiency+suffer+only+intermittent+haemolysis.+The+blood+film+then+shows+irregularly+contracted+cells+%5Bdeep+red+arrows%5D+and+sometimes+hemighosts+%5Bdeep+blue+arrow%5D+in+which+all+the+haemoglobin+appears+to+have+retracted+to+one+side+of+the+erythrocyte..jpg "It has an X-linked recessive inheritance so occurs mainly in males. In high incidence areas female homozygotes occur and have the same features as male hemizygotes. Haemolysis is precipitated by infection and exposure to exogenous oxidants such as broad beans, naphthalene and certain drugs. Between acute haemolytic episodes the blood film is normal.")
38
Blood film in glucose-6-phosphate dehydrogenase (G6PD) deficiency showing a hemighost [red arrow] and a keratocyte [blue arrow]. Keratocytes are formed when a Heinz body is removed from an erythrocyte by the spleen. During acute haemolysis a Heinz body preparation is positive.
![Blood film in glucose-6-phosphate dehydrogenase (G6PD) deficiency showing a hemighost [red arrow] and a keratocyte [blue arrow].](http://slideplayer.nl/slide/2028533/8/images/38/Blood+film+in+glucose-6-phosphate+dehydrogenase+%28G6PD%29+deficiency+showing+a+hemighost+%5Bred+arrow%5D+and+a+keratocyte+%5Bblue+arrow%5D..jpg "Keratocytes are formed when a Heinz body is removed from an erythrocyte by the spleen. During acute haemolysis a Heinz body preparation is positive..")
39
Macrocytaire anemie “Fout” macrocytair - koude agglutininen
- diabetes mellitus “Echt” macrocytair - reticulocytosis - verstoorde DNA synthese - leverlijden - medicatie - CDA type I en III

40
koude agglutininen

41
Screening bij vermoeden van een abnormale bloeding
PBO en uitstrijkje PT APTT Fibrinogeen Trombinetijd (reptilasetijd) Bloedingstijd
 Bloedingstijd.")
42
Wat vind je niet met deze screening?
Trombocytopatieën zonder morfologische afwijkingen Matige deficiënties van stollingsfactoren (tot ± 50%) (normaal ook geen klinische problemen) Een aantal vormen van vWD Een minderheid van patiënten met lupus anticoagulans FXIII deficiëntie (Verhoogde tromboseneiging)
 (normaal ook geen klinische problemen) Een aantal vormen van vWD. Een minderheid van patiënten met lupus anticoagulans. FXIII deficiëntie. (Verhoogde tromboseneiging)")
43
Fouten bij de hemostatische screening
Resultaat Oorzaak Hoe herkennen? correctie Laag aant.blpl in teller Stolsel Plaatjesaggl in vitro EDTA-afh. aggl. Satellitisme (EDTA) Te grote blpl (BS) Visuele inspectie uitstrijkje Uitstrijkje Opnieuw Citraat manueel ↑ aant. Blpl in teller RBCfragmenten WBCfragmenten Manueel ↑ PT of APTT Bewaring staal > 4h ↑antistolling door plasmadeficiet Fgn lipemie Traceren Polycytemie (Hct> 55%), of te weinig staal Fgn < 80 mg/dL Visueel Opnieuw afnemen Citraat conc aanpassen (3.8%) x(100-Hct%)xvol bloed =mL citraat 3.8%
 Te grote blpl (BS) Visuele inspectie. uitstrijkje. Uitstrijkje. Opnieuw. Citraat. manueel. ↑ aant. Blpl in teller. RBCfragmenten. WBCfragmenten. Manueel. ↑ PT of APTT. Bewaring staal > 4h. ↑antistolling door plasmadeficiet. Fgn lipemie. Traceren. Polycytemie (Hct> 55%), of te weinig staal. Fgn < 80 mg/dL. Visueel. Opnieuw afnemen. Citraat conc aanpassen (3.8%) x(100-Hct%)xvol bloed. =mL citraat 3.8%")
44
Fouten bij de hemostatische screening
Resultaat Oorzaak Hoe herkennen? correctie Fgn , onstolbaar bloed Stolsel Visueel Opnieuw afnemen Fgn (automaat) Lipemie manueel Geen stolling in stoltesten Heparine contaminatie Overleg met kliniek Onverwachte stolling Hypercalcemie (toegediend Ca) Ca toediening en opnieuw
 Lipemie. manueel. Geen stolling in stoltesten. Heparine contaminatie. Overleg met kliniek. Onverwachte stolling. Hypercalcemie (toegediend Ca) Ca toediening en opnieuw.")
45
Bijzonderheden in de pediatrische stollingsanalysen
Zie de referentiewaarden Stollingscomponenten gaan niet door de placentabarrière, daarom kunnen een aantal deficiënties bij het jonge kind niet betrouwbaar opgespoord worden: Matige deficiëntie van F IX (= normaal laag) De meeste gevallen van vWD (vWF normaal hoog) Heterozygote deficiënties van de inhibitoren (C, S, AT; normaal laag) Bij het (algemeen) zieke kind kunnen sommige waarden lager zijn dan verwacht Bloedplaatjestelling is niet erg verschillend, maar bij aggregatietesten zijn normaal verstoord voor ADP, adrenaline en collageen
 De meeste gevallen van vWD (vWF normaal hoog) Heterozygote deficiënties van de inhibitoren (C, S, AT; normaal laag) Bij het (algemeen) zieke kind kunnen sommige waarden lager zijn dan verwacht. Bloedplaatjestelling is niet erg verschillend, maar bij aggregatietesten zijn normaal verstoord voor ADP, adrenaline en collageen.")
46
Referentiewaarden: stollingsanalysen
Prematuur (d1) A term (d1) Volwassene maturatietijd PT, s APTT, s 3 maand Fgn, mg/dL II 6 maand V 5 dagen VII VIII:C vWF:Ag IX 6-9 maand X XI XII XIII Plgn DD Kan ↑ Enkele dagen
 A term (d1) Volwassene. maturatietijd. PT, s APTT, s maand. Fgn, mg/dL II maand. V dagen. VII VIII:C vWF:Ag IX maand. X XI XII XIII Plgn DD. Kan ↑ Enkele dagen.")
47
Referentiewaarden: stollingsinhibitoren
Analyse Prematuur, d1 A term, d1 Volwassen maturatietijd AT (IU/mL) 6 maand PC (IU/mL) > 6 maand PS totaal* 6-12 maand * Vrij PS is relatief meer aanwezig omwille van lage C4 BP spiegels
 maand. PC (IU/mL) > 6 maand. PS totaal* maand. * Vrij PS is relatief meer aanwezig omwille van lage C4 BP spiegels.")
48
Bijzonderheden in de pediatrische stollingsanalysen
Zie de referentiewaarden Stollingscomponenten gaan niet door de placentabarrière, daarom kunnen een aantal deficiënties bij het jonge kind niet betrouwbaar opgespoord worden: Matige deficiëntie van F IX (= normaal laag) De meeste gevallen van vWD (vWF normaal hoog) Heterozygote deficiënties van de inhibitoren (C, S, AT; normaal laag) Bij het (algemeen) zieke kind kunnen sommige waarden lager zijn dan verwacht Bloedplaatjestelling is niet erg verschillend, maar aggregatietesten zijn normaal verstoord bij neonati voor ADP, adrenaline en collageen
 De meeste gevallen van vWD (vWF normaal hoog) Heterozygote deficiënties van de inhibitoren (C, S, AT; normaal laag) Bij het (algemeen) zieke kind kunnen sommige waarden lager zijn dan verwacht. Bloedplaatjestelling is niet erg verschillend, maar aggregatietesten zijn normaal verstoord bij neonati voor ADP, adrenaline en collageen.")
49
Small bruises and bilateral haemorrhage into the knee joints (haemarthroses) in haemophilia A.
 in haemophilia A.")
50
The descendants of Queen Victoria, showing the inheritance of haemophilia A. Because inheritance is sex-linked recessive the great majority of sufferers are male whereas asymptomatic carriers are female.

51
Stollingsanalysen ivm de differentiaal diagnose
vWD en hemofilie A Analyse Hemofilie A Von Willebrand PT Normaal normaal APTT Verlengd verlengd Trombine tijd FVIII Gedaald Gedaald of nl vWF:Ag vWF:Rco blplaggregatie ristocetine of nl Bloedingstijd Verlengd of nl

52
Andere oorzaken voor een verlengde APTT
Lupus anticoagulans (geen of onvolledige correctie bij mix-experimenten met normaal plasma ; wel correctie door toevoegen van fosfolipiden) Meest frequent sec op virale infecties Bij de neonaat + vanuit de moederlijke circulatie via placenta Heparine F IX deficiëntie (slechts 10 – 20 % van hemofilie A) Overerving cfr hemofilie A Carriers hebben ook een verlaagd F IX en zijn frekwenter symptomatisch dan bij F VIII deficiëntie De verworven vorm (inhibitoren) (onmiddellijke inhibitie) Zeldzaam: F XII deficiëntie, PK def., HMWK def, F XI def.
 Meest frequent sec op virale infecties. Bij de neonaat + vanuit de moederlijke circulatie via placenta. Heparine. F IX deficiëntie (slechts 10 – 20 % van hemofilie A) Overerving cfr hemofilie A. Carriers hebben ook een verlaagd F IX en zijn frekwenter symptomatisch dan bij F VIII deficiëntie. De verworven vorm (inhibitoren) (onmiddellijke inhibitie) Zeldzaam: F XII deficiëntie, PK def., HMWK def, F XI def.")
53
Verlengde PT & APTT Te veel citraat Lupus anticoagulans
Deficiëntie van het protrombine-complex (II, VII, IX, X) (PT meestal meer verlengd dan APTT) Confirmatie door factorbepaling Indien vit K def. is er een correctie na ± 12 h door vit K toediening Vit K def. tgv verminderde inname of een verminderde leverfunctie (fysiologisch bij de neonaat)
 (PT meestal meer verlengd dan APTT) Confirmatie door factorbepaling. Indien vit K def. is er een correctie na ± 12 h door vit K toediening. Vit K def. tgv verminderde inname of een verminderde leverfunctie (fysiologisch bij de neonaat)")
54
Trombocytopenie op kinderleeftijd wellicht meest frequent probleem
Megakaryocyten (gecombineerd met pancytopenie bij Fanconi met eerst de trombopenie), TAR (geïsoleerde trombopenie) Ineffectieve trombopoiese (Bernard Soulier, May-Hegglin,…) Redistributie Hypersplenisme (meestal > /µL) Verhoogd verbruik DIC Macroscopische trombose Verhoogde afbraak Immuungemedieerd (meest frequent) Niet-immuun : extrinsiek intrinsiek (trombocytopatie)
, TAR (geïsoleerde trombopenie) Ineffectieve trombopoiese (Bernard Soulier, May-Hegglin,…) Redistributie. Hypersplenisme (meestal > /µL) Verhoogd verbruik. DIC. Macroscopische trombose. Verhoogde afbraak. Immuungemedieerd (meest frequent) Niet-immuun : extrinsiek. intrinsiek (trombocytopatie)")
55
Immuungemedieerde trombocytopenie
Auto-immuun -postviraal (acute ITP) (varicella, rubella, EBV, hepatitis, HIV, CMV, of respiratoire of gastro-intestinale virussen). De trombocytopenie treedt op 1 a 6 weken na de infectie, en stelt zich snel in (binding van Ag/Ab complexen), ¾ herstelt binnen 3 maand Dikwijls < /µL. Piekincidentie bij kinderen tussen 2 en 4 jaar oud -niet-viraal ; bacterieel geïnduceerde Ig-binding van bloedplaatjes -chronische ITP (bij oudere kinderen (> 5 jaar), trage ontwikkeling, spontaan herstel 1/3, herstelperiode > 1 jaar) -geassocieerd met een auto-immune aandoening (SLE, RA) of medicatie (zeldzaam) Allo-immuun fetomaternale incompatibiliteit of bij transfusies
 (varicella, rubella, EBV, hepatitis, HIV, CMV, of respiratoire of gastro-intestinale virussen). De trombocytopenie treedt op 1 a 6 weken na de infectie, en stelt zich snel in (binding van Ag/Ab complexen), ¾ herstelt binnen 3 maand. Dikwijls < /µL. Piekincidentie bij kinderen tussen 2 en 4 jaar oud. -niet-viraal ; bacterieel geïnduceerde Ig-binding van bloedplaatjes. -chronische ITP (bij oudere kinderen (> 5 jaar), trage ontwikkeling, spontaan herstel 1/3, herstelperiode > 1 jaar) -geassocieerd met een auto-immune aandoening (SLE, RA) of medicatie (zeldzaam) Allo-immuun. fetomaternale incompatibiliteit of bij transfusies.")
56
Trombocytopenie op kinderleeftijd Onderzoeken
Geassocieerde ziektebeelden (eczema (WAS), nieraandoeningen, doofheid, familiaal voorkomen) Bloedplaatjesmorfologie Beenmergonderzoek : aantal en morfologie Bloedplaatjesfunctie (indien trombocytopatie vermoed bij te excessieve bloeding relatief tov het aantal plaatjes) Bloedplaatjesantilichamen (?) Eventueel meer gespecialiseerde testen indien nodig
, nieraandoeningen, doofheid, familiaal voorkomen) Bloedplaatjesmorfologie. Beenmergonderzoek : aantal en morfologie. Bloedplaatjesfunctie (indien trombocytopatie vermoed bij te excessieve bloeding relatief tov het aantal plaatjes) Bloedplaatjesantilichamen ( ) Eventueel meer gespecialiseerde testen indien nodig.")
58
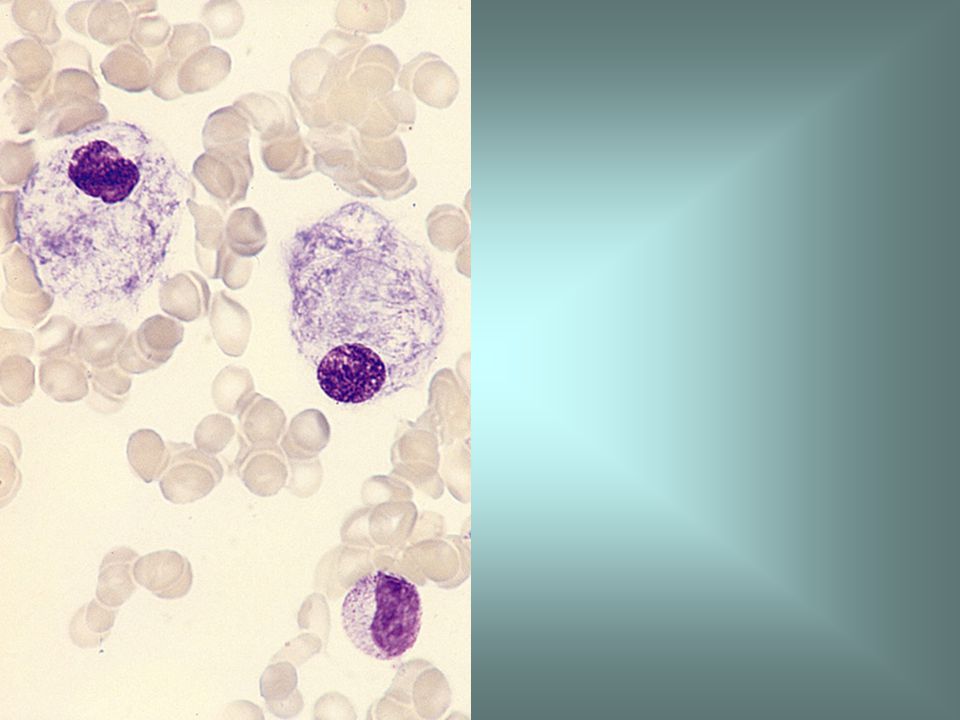
Bone marrow aspirate in Gaucher's disease or hereditary glucosyl ceramide lipidosis, showing Gaucher's cells. Gaucher's disease is an inherited metabolic defect. Gaucher's cells are altered macrophages containing glucocerebroside, the metabolite which accumulates in this disease.

60
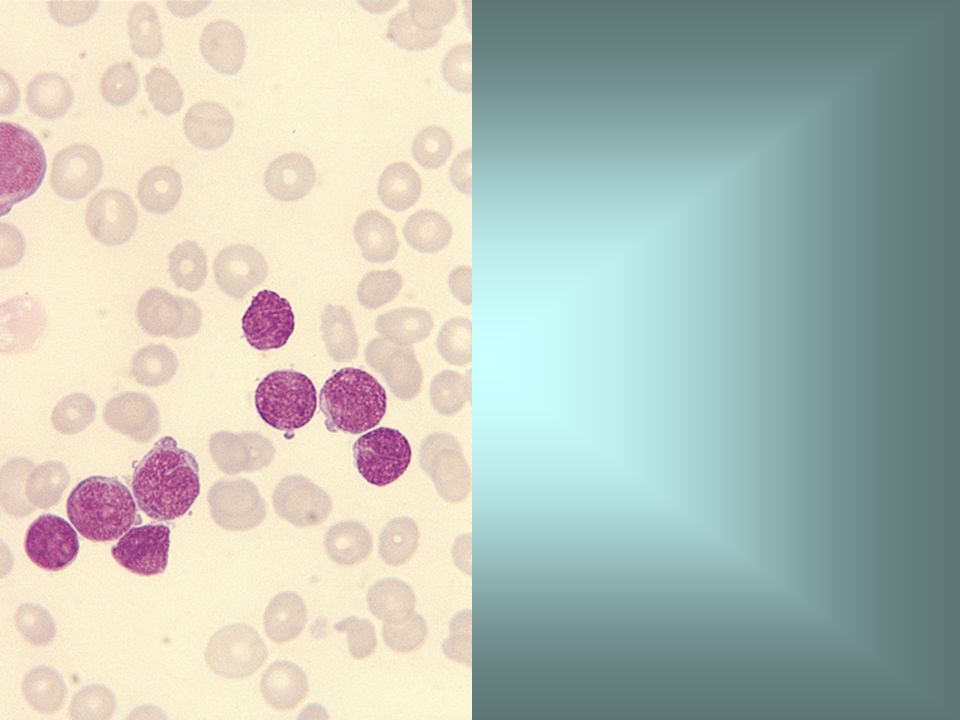
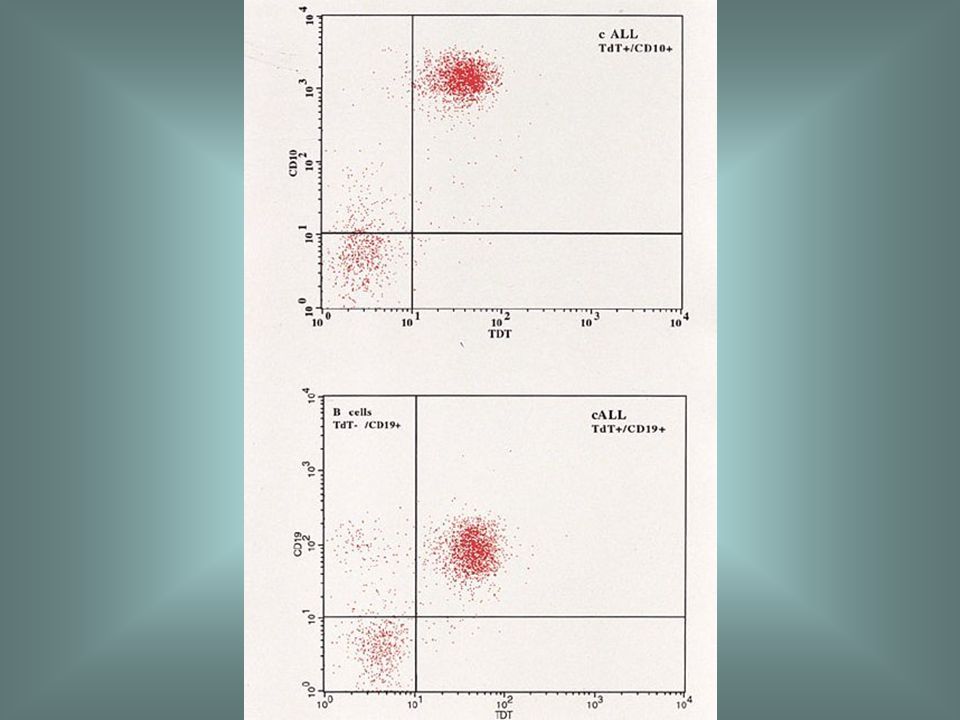
Precursor B ALL Acute lymphoblastic leukaemia, either B or T lineage.
The peak incidence is in early childhood, particularly between the ages of 2 and 10 years. Common clinical features are bruising, pallor, bone pain, lymphadenopathy, hepatomegaly and splenomegaly. In T-lineage cases a chest X-ray may show enlargement of the thymus. A morphological classification into L1, L2 and L3 categories has been proposed by the FAB group. ALL can be further classified on the basis of either cytology, immunophenotype or cytogenetic and molecular genetic features.

63
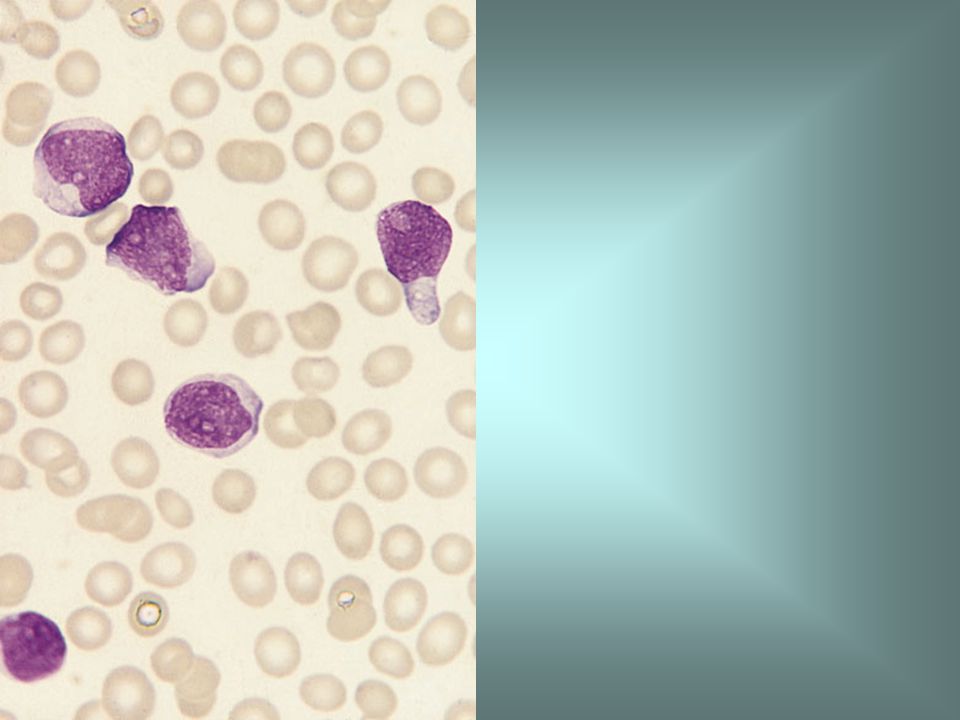
Blood film in L2 ALL showing pleomorphic medium to large blasts with small but distinct nucleoli.

65
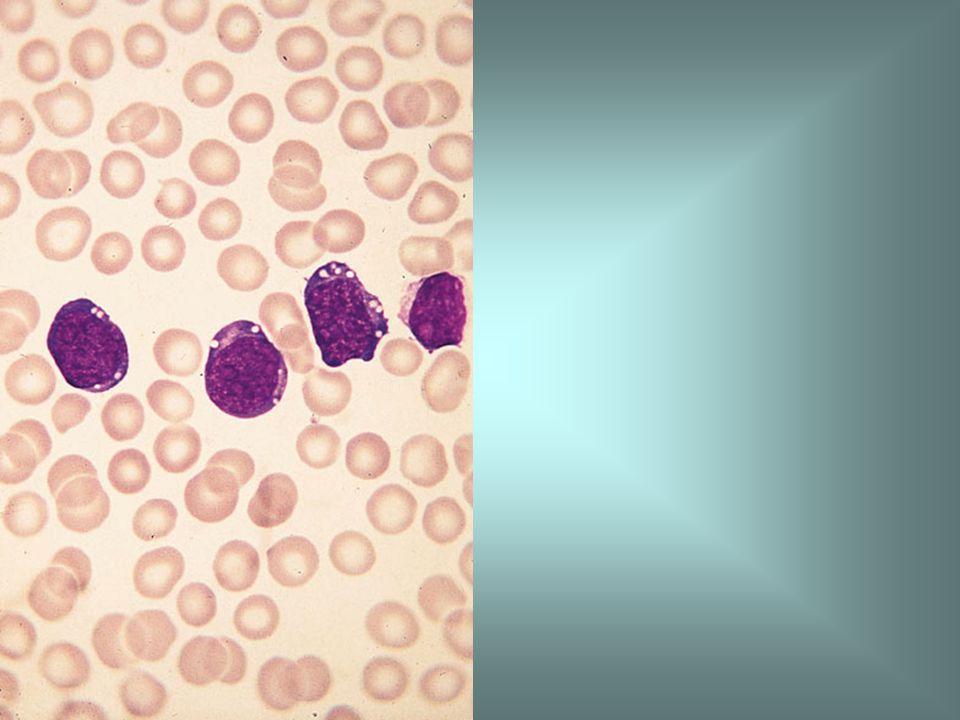
Blood film in L3 ALL showing strong cytoplasmic basophilia and cytoplasmic vacuolation.

67
Normal immature lymphoid cells, referred to as haematogones, in the bone marrow of a child with medulloblastoma.

69
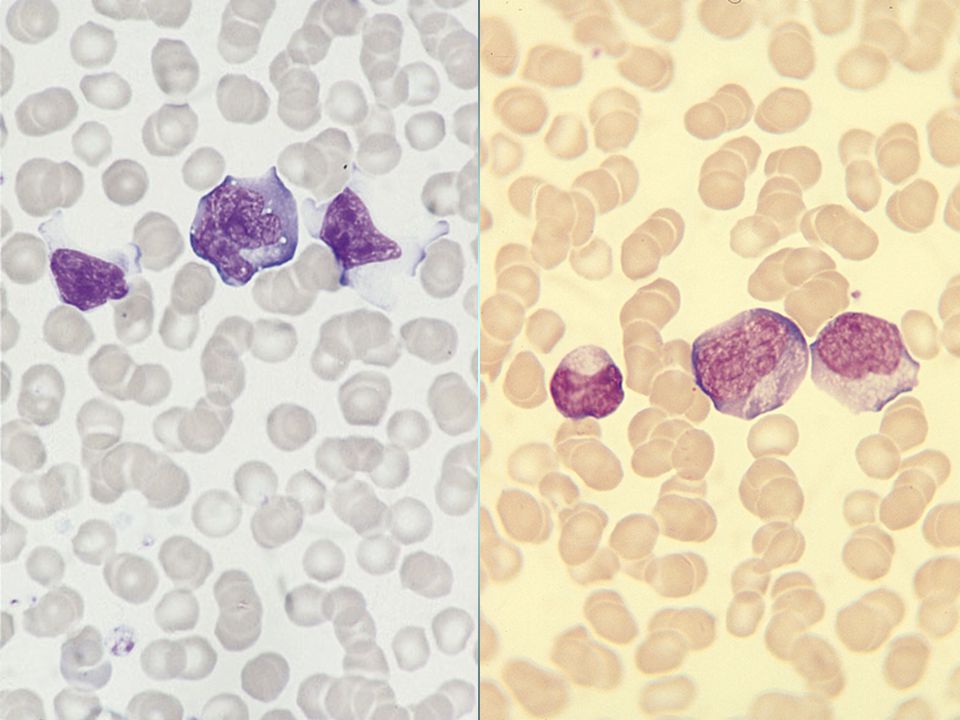
Reactieve lymfocyten

Verwante presentaties










![Deltion College Engels C1 Gesprekken voeren [Edu/002]/ subvaardigheid lezen thema: Order, order…. can-do : kan een bijeenkomst voorzitten © Anne Beeker.](/8/2048322/big_thumb.jpg "Deltion College Engels C1 Gesprekken voeren [Edu/002]/ subvaardigheid lezen thema: Order, order…. can-do : kan een bijeenkomst voorzitten © Anne Beeker.>")



to watch throughout the month of August,>")
 Quiz Night !>")




